I like this project because it starts with a classifier question and then refuses to stay only a classifier question.
The setup is TCGA BRCA, PAM50 subtype labels, and a mix of clinical, RNA, methylation, epigenetic age, and immune-context features. A plain machine-learning version of the project would ask whether those tables can predict subtype. The more interesting version asks what the useful features might be saying biologically.
The aging piece comes from DNA methylation clocks. A clock estimates biological age from methylation patterns, but in cancer the useful signal is often the gap between methylation age and chronological age. That delta-age value asks whether a tumor looks older or younger than expected.
The report compared Horvath, Hannum, and PhenoAge clocks across subtype groups. The pattern I found most interesting was subtype-specific: Luminal tumors tended to show positive age acceleration, while Basal tumors sat closer to zero or negative delta-age.
Hannum looked strongest in the downstream linear model, with an approximate R-squared around 0.30 in the report. I would not oversell that number, but it was enough to make the aging signal worth carrying into the rest of the analysis.
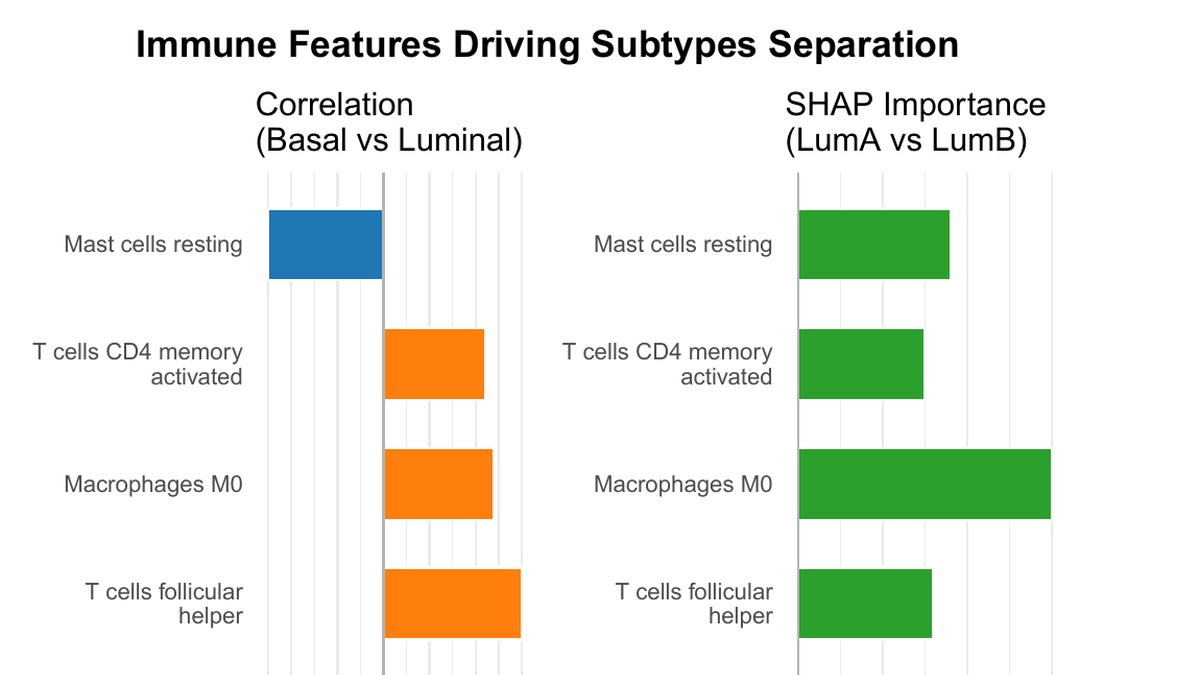
The immune layer made the story less generic. Resting mast cells and M0 macrophages appeared as strong delta-age correlates, suggesting that some age-accelerated tumors may be carrying an innate or wound-healing-like immune profile. Follicular helper T cells and macrophage patterns also helped separate Luminal and Basal structure.
That is the part I care about more than raw accuracy. If a model separates subtype groups, the next question is whether the features point back to plausible biology or only to technical shortcuts.
The repo includes notebooks for metadata cleaning, RNA preprocessing, methylation preprocessing, delta-age calculation, immune-score integration, classification, and annotation. It also preserves report artifacts for the epigenetic age statistics and downstream annotation.
I would still frame this as an applied multi-omics analysis, not a clinical classifier. The next version should clean up regeneration paths, remove absolute report paths, and make train-test reporting and feature-group sensitivity analysis easier to inspect.
References and artifacts: the analysis is in the BRCA multi-omics repository.