The simple version of this project was: can a protein language model look at APP variants and recover any useful signal without being trained on the experiment?
The more honest version is slightly messier. APP is biologically important, the ProteinGym A4_HUMAN_Seuma_2021 dataset gives a real deep-mutational scanning benchmark, and ESM models give several possible ways to score mutations. The question is not only whether ESM works, but which scoring formulation is doing the work.
The dataset has roughly 14,483 APP variants, including single and double mutants. That makes it useful for looking beyond a clean one-mutation-at-a-time story.
I compared ESM-1v and ESM-2 scores using masked log-likelihood ratios, pseudo-log-likelihood, embedding distance from wild type, entropy-weighted masked scores, ensemble-style masked scores, and mutant marginal probability.
The important thing is that “an ESM score” is not one object. A masked score, a pseudo-likelihood score, and an embedding-distance score can all come from the same broad model family but behave differently on the same variants.
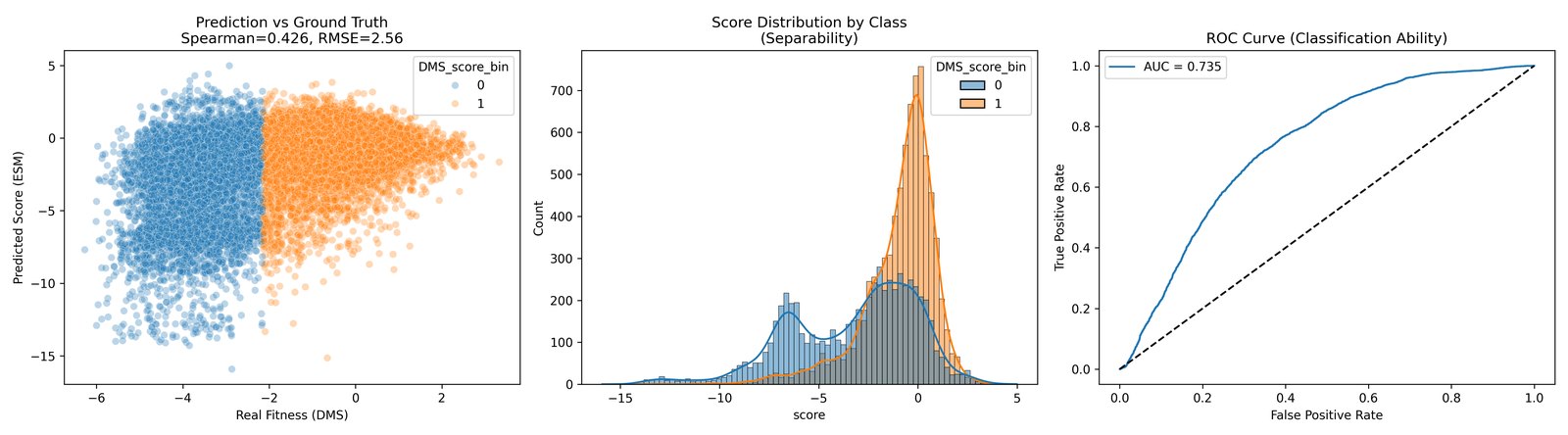
The strongest APP runs landed around the low-to-mid 0.4 range in Spearman correlation against the experimental DMS scores. For example, ESM2 650M pseudo-log-likelihood and ensemble masked-score runs were both in that neighborhood, with top-100 precision in the low teens.
That is not a magic result, but it is also not nothing. I read it as partial ranking signal: useful for prioritization, not sufficient for mechanistic explanation.
The double-mutant side is where the project becomes more interesting. Additive thinking can break down when two substitutions interact, and language-model scores sometimes pick up context that simpler mutation penalties miss. That does not make the model causal, but it makes the score worth interrogating.
What I would not claim is that ESM “understands” APP biology. A moderate correlation can hide all kinds of local failures. It can rank some variants well while missing the biological reason they matter.
The next version should make the ablation cleaner: model size, score type, variant class, and evaluation metric separated in one table. That would make it easier to see whether the useful signal is coming from scale, scoring, or the structure of the benchmark itself.
References and artifacts: the notebooks, figures, scripts, and result tables are in the ESM-DeepLearning repository.